|
|
|
|||||
|
||||||
MAH试点、进入ICH、关联申报:倒逼原料药企升级提速摘要:
医药网5月2日讯 在新政策环境及其产生的连锁效应下,原料药企的全面升级迫在眉睫。
2017年11月30日,原CFDA关于调整原料药、药用辅料和药包材审评审批事项的公告(2017年第146号),要求有关企业或者单位可通过登记平台按公告要求提交原料药、药用辅料和药包材登记资料,获得原料药、药用辅料和药包材登记号,待关联药品制剂提出注册申请后一并审评。公告发布前已获得批准文号的原料药、药用辅料和药包材相关登记要求,将在登记平台建立后另行通知。
在登记平台建立的过渡期,药审中心在门户网站以表格方式对社会公示“原料药登记数据”“药用辅料登记数据”“药包材登记数据”。以原料药为例,原料药登记资料主要内容包括基本信息、生产信息、特性鉴定、原料药的质量控制、对照品、药包材、稳定性等。
同年12月4日,原CFDA办公厅公开征求《原料药、药用辅料及药包材与药品制剂共同审评审批管理办法(征求意见稿)》意见(以下简称“征求意见稿”)。这意味着原料药、药用辅料及药包材(以下简称“原辅包”)关联审评审批制度的序幕即将拉开。
上市许可持有人责任大
[趋势] 制剂质控从严,对原辅包企提出新要求
根据“征求意见稿”,药品上市许可持有人承担制剂质量的主体责任,建立以制剂为核心、以原辅包为基础的质量管理体系。这就要求药品制剂上市许可持有人建立的质量管理体系能涵盖制剂全生命周期的质量管理,对制剂所用的原辅包质量能够有效追溯,文件管理中必须要明晰原辅包来源、批次、生产、质控和变更情况。
药品制剂上市许可持有人需要和原辅包企业一同签署“原辅包企业保证持续稳定地供应符合制剂质量的原辅包产品的长期供货/质量保证协议”,原辅包企业提交必要信息以便评估和控制由原辅包引入制剂的质量风险。药品制剂与原辅包不是同一申请人的,药品制剂申请人的申报资料还要提供原辅包上市许可持有人或者企业的授权使用书,这要求药品制剂上市许可持有人在原辅包的关联合同中要有相关诉求。
药品上市许可持有人还要对原辅包开展供应商审计,从制剂全生命周期的质量管理新理念出发,审计将要在研发阶段前启动。审计的内容一般包括现场GMP考核、工厂厂商资质审核和质量体系文件审核、产品注册文件和年度报告。值得关注的是,研发阶段一些辅料所需的供应量比较低,辅料供应商未必愿意接受审计;此外,如果原辅包企业觉得药品上市许可持有人有盗取资料的风险,也有可能会拒绝合作。
随着一致性评价法规对制剂的体外溶出速率、生物利用度和稳定性要求不断提高,粒径大小若是影响溶出度、溶解度、生物利用度,制剂生产、稳定性、含量均匀度,以及产品外观的关键因素,通常都要制定粒径限度要求。在这种情况下,国内的制剂企业通常选择将压力往上游传递,要求原料药生产企业加强粒径控制,特别是直接混合原辅料压片工艺制剂对应的原料药,以往的过筛控制已不能满足制剂生产企业的需求。
此外,根据“征求意见稿”,同一原料药生产企业供不同给药途径制剂使用且质量存在差别的同一原料药,应当按不同登记号登记;给药途径相同、生产工艺相近,仅晶型、粒径等质控要求不同的原料药,应当在同一登记号下对不同工艺、晶型、粒径进行分类并编号。这将有利于原料药生产企业供货不同粒径控制需求的制剂企业。
ICH法规受注重
[趋势] 倒逼合作原料药厂质量升级
中国进入ICH后,原料药也要参考ICH原料药开发和制造管理原则。目前CDE的法规与规章栏目“ICH指导原则”的质量指导原则已发布了不少原料药相关的中文版,如《Q1A(R2):新型原料药和药品的稳定性测试》《Q1B: 稳定性测试: 新型原料药和药品的光稳定性测试》《Q1D:新型原料药和药品稳定性测试的交叉和矩阵设计》《Q3A(R2): 新型原料药中的杂质问题》《Q6A: 质量规格:新原料药和药品的检验程序和可接收标准:化学物质》和《Q7: 原料药GMP指南》等与原料药相关的指导原则。
ICH制度影响最大的是国内已有批文的原料药厂家,下游制剂生产企业一致性评价质量提升压力,将倒逼已合作的原料药厂家的质量体系进行升级。例如杂质谱的全面研究,包括降解杂质、遗传毒性杂质和元素杂质的研究;残留溶剂中有无应避免的溶剂,应限制的溶剂又无超过规定 PDE(the Permitted Daily Exposure,允许日接触量);起始物料的审计与管理制度等。
关联申报“试用期”
[趋势] 欧美原料供货商将回流
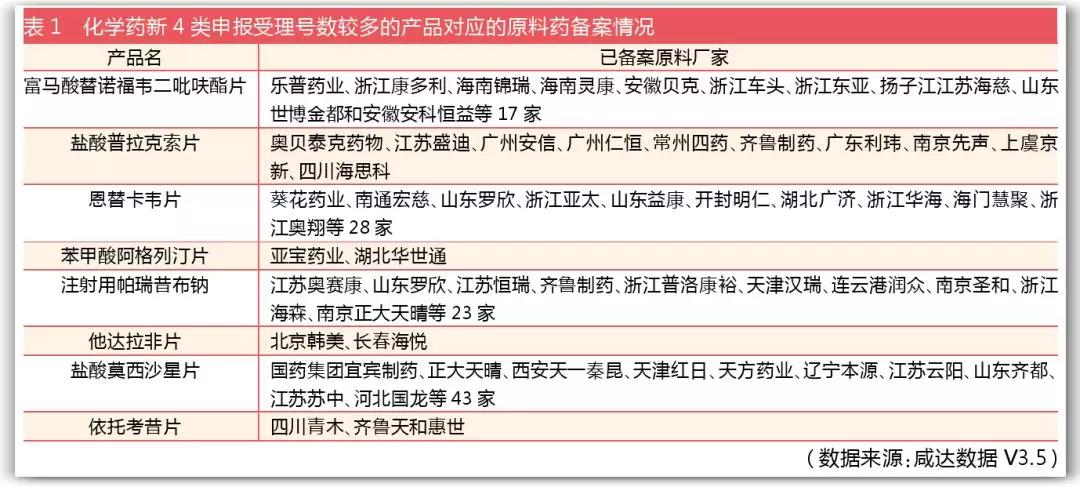
截至2018年4月23日,共有2037条原料药信息备案。咸达数据V3.5整理了化学药新注册分类办法4类的申报受理号数较多的产品对应的原料药厂家备案情况后发现,除了富马酸替诺福韦二吡呋酯片原研厂家香港吉立亚科学有限公司备案的原料药状态为“已有上市制剂使用该原料”,新4类的其他制剂对应的原料药暂无“单独申报且已通过技术审评的原料,尚未与制剂进行共同审评”或“已登记但尚未通过技术审评的原料,正在与关联制剂共同审评”状态,个别产品为“已登记但尚未通过技术审评的原料,尚无关联制剂申报”状态。由此可见,原料药和制剂的关联情况暂不能从数据库中获悉。
一直致力于欧美市场的中国原料药生产厂家,以往由于价格和注册法规的原因,绝少供货国内生产厂家。关联审评政策出台后,由于一致性评价的刚性需求,这些企业有可能回流国内参与原料药市场的竞争。制剂企业在选择此类企业的时候,需要了解过往现场核查中主要发现了哪些缺陷,还要了解国内外注册法规要求的不同点,例如国内原料药CTD文件要求原料药也要做包材相容性的研究。
为了保证一致性评价的注册进度,制剂企业选择原料药厂家时,会选择在美国有DMF、欧盟CEP申请,并且通过欧美GMP认证和现场核查的生产厂家。目前供货的主要有欧洲、印度和中国的生产企业。
过了一致性评价后,制剂上市许可持有人更换原料药生产企业是否还需要重新做生物等效性试验法规暂未明确。根据欧美的法规,制剂上市许可持有人根据其对原料药的标准重新选择经济性更好的原料药企业只需要做三批验证即可。关联申报制度下,原料药备案平台中“已有上市制剂使用该原料”对应的非原研厂家的原料药生产商就可以参与原料药供应竞争,以往国内由于生产批件所造成的原料药垄断情形会减少,相应地,技术壁垒和专利所导致的市场供应不足将会越来越多出现。
 |
|
Copyright ©2015 王中王中特
粤ICP备15022662号(粤)
-技术支持:信息管理部|联系我们
|